

What Is The Difference Between A Regular 13c Nmr And A 13c Nmr Registered In A Proton Coupled Mode?
![]() Open access peer-reviewed affiliate
Open access peer-reviewed affiliate
1H and 13C NMR for the Profiling of Natural Product Extracts: Theory and Applications
Submitted: June 2nd, 2017 Reviewed: September 18th, 2017 Published: Dec 6th, 2017
DOI: 10.5772/intechopen.71040

IntechOpen Downloads
ii,189
Total Chapter Downloads on intechopen.com
Abstract
Nuclear magnetic resonance (NMR) spectroscopy and mass spectrometry (MS) are the primary methods of metabolomics, the branch of '-omics' that deals with small molecules. Although MS is gaining popularity in metabolomics, NMR enjoys a number of primal advantages because information technology is nondestructive, unbiased, quantitative, does not require separation or derivatization, and is amenable to compounds that are hard to analyze past gas chromatography-mass spectrometry (GC-MS) and liquid chromatography-mass spectrometry (LC-MS). At that place are two general approaches to the employ of NMR for profiling studies: an untargeted arroyo, which uses chemometric analysis; and a targeted approach, which aims to quantify known compounds in the extract. These approaches, yet, are not mutually exclusive and volition likely converge in the hereafter. This paper volition draw the basic theoretical principles that should be considered to develop NMR into a standard quantitative method. Although 1H NMR is more sensitive, 13C NMR spectra are simpler with less overlapping signals and are less affected by different magnetic field strengths. Diverse applications of 1H and 13C NMR for the profiling of natural products are described. The use of two-dimensional 1H NMR has been used to overcome issues of spectral overlap. The standardization of the NMR protocol will make it a more useful tool for the profiling of natural products extracts.
Keywords
- nuclear magnetic resonance
- 1H NMR
- 13C NMR
- natural products profiling
- metabolomics
- chemometrics
*Address all correspondence to: fdayrit@ateneo.edu
i. Introduction
The objective of this paper is to review the applications of 1H and 13C nuclear magnetic resonance (NMR) for the quantitative profiling of plant natural products extracts and the theoretical parameters that should exist considered, if information technology is to become a more useful tool.
NMR and mass spectrometry (MS) are the principal methods of metabolomics, the branch of '-omics' that deals with small molecules. The Metabolomics Society describes metabolomics as: "the comprehensive label of the pocket-sized molecule metabolites in biological systems" [1]. NMR has a number of characteristics that meet the requirements of metabolomics: it is authentic, quantitative, comprehensive, unbiased, and is able to provide data that can be used to make up one's mind molecular structure. The review will talk over these aspects in item.
1.1. NMR and MS
Although MS techniques, such as gas chromatography-mass spectrometry (GC-MS) and liquid chromatography-mass spectrometry (LC-MS), are about usually used in metabolomics, NMR still enjoys a number of key advantages. In particular, NMR is nondestructive, unbiased, quantitative, does not crave separation or derivatization, and is acquiescent to compounds that are difficult to clarify by GC-MS and LC-MS. For example, GC-MS often requires derivatization of compounds, such as sugars and amines. LC-MS, on the other hand, mostly requires sample preparation, chromatographic separation, specific experimental and ionization conditions, instrumentation and operator skill [2]. These make information technology difficult to standardize MS assay. In contrast, NMR does not require elaborate sample training and fractionation, is highly reproducible, and is able to provide both qualitative and quantitative data on chemically diverse compounds [3, 4]. The standardization of the NMR protocol will further amend the usefulness of NMR equally a tool for the profiling of natural products extracts. Because NMR is able to find compounds only down to 0.1% level, it is not suitable for the detection of trace components. NMR is less sensitive than MS, which can observe compounds downwardly to parts per million (ppm) levels. Because of the distinct advantages of each method, NMR and MS are considered equally complementary techniques.
NMR is a quantitative spectroscopic tool because the intensity of the peaks is directly proportional to the number of nuclei. With improvements in electronics and the use of higher magnetic field strengths, the sensitivity and resolving ability of NMR has improved. Nevertheless, the lack of standardized protocols has express its quantitative application and many consider NMR mainly as a qualitative method, mainly for chemic structure determination and molecular dynamics [5].
The employ of NMR as a quantitative method has been expanding, giving rise to the term "quantitative NMR" (qNMR). The pharmaceutical industry, which has stringent requirements of analysis, has been turning to the utilize of qNMR in early drug evolution to address the need for rapid, selective, and authentic assay without requiring expensive and tedious chromatographic methods. It is too worth noting that qNMR meets the stringent regulatory standards of the pharmaceutical industry, including the International Briefing on Harmonization. qNMR has been applied mainly to 1H nuclei although 19F and 31P NMR have also been used where appropriate considering of their 100% isotopic abundance [6]. The chief advantages of qNMR are its accuracy, reproducibility, and flexibility with respect to the nature of the analyte, the only requirement being the presence of protons and carbon, and its ability to simultaneously quantify multiple analytes, particularly when validated using external scale. Quantitative 1H NMR (qHNMR) has been shown to have an accuracy and precision of ±1% and an uncertainty of measurement of less than 0.1%. This makes it suitable as a metrological technique for the certification of purity of organic compounds [seven].
There are ii full general approaches to the use of NMR for profiling or metabolomics studies. In the beginning approach, only the spectral patterns (chemical shifts and intensities) are recorded and are used to compare and group samples. In this arroyo, compounds are non initially identified. Considering statistical tools, such as master components analysis (PCA) are used, this is sometimes called a chemometric arroyo. In the second arroyo, particular compounds which are known to be present in the extract are identified and quantified using a reference spectral library. This approach is referred to as quantitative or targeted metabolomics [8]. These approaches, notwithstanding, are not mutually sectional and volition likely converge in the futurity with improved statistical tools and bigger NMR spectral databases.
Because of the large amount of data that are produced, statistical methods, known equally chemometrics, are applied to reduce the number of variables. Chemometrics is a family of statistical techniques that are practical to large sets of chemical data, such as NMR chemic shift peaks, with the objective of gaining insights into the characteristics of the samples through the utilise of graphical representation [9]. Considering chemometrics is able to process large amounts of information, it is an ideal tool for NMR which produces a lot of data (chemic shifts). This can exist used to notice patterns of groupings and correlations among natural production samples which can exist used for quality control and standardization [x]. Since chemometrics started to exist practical to NMR around the year 2000, progress has been very rapid. Chemometrics has been used to classify whole constitute samples based on their NMR profiles according to species, origin, processing treatment, age, and various quality parameters [xi].
Advertisement
2. 1H and 13C NMR as profiling methods
In a talk given during the William Draper Harkins Lecture, University of Chicago in 1991, Alexander Pines mentioned that his organic chemical science colleagues at Berkeley consider two vital instruments in a research laboratory: a residue and an NMR spectrometer. His view is not surprising as decades of improvement in both instrumentation and techniques had rendered the NMR spectrometer every bit a tool of pick in characterizing molecules, from the structures of natural products and constructed organic compounds to biomolecules and organometallic complexes. NMR spectroscopy takes advantage of the interaction betwixt nuclei that are acting as tiny magnets and an external magnetic field and this provides a powerful means of probing the chemical bonding and environment of the nucleus. These phenomena are key to the applicability of 1H and 13C NMR to natural products.
2.1. 1H NMR spectroscopy
Hydrogen is present in virtually every organic molecule, and its major isotope, 1H, has an abundance of 98.985%. The 1H nucleus reports a frequency specific to its immediate vicinity in an NMR spectrum. This frequency is extremely sensitive to the electronic environs thus giving each 1H nucleus in an organic compound a type of identification number, called the NMR chemic shift. Magnetic nuclei, such as 1H, also interact with each other. In solution or liquid-state NMR spectroscopy, these interactions, chosen couplings, are observed as "splitting" of lines in an NMR spectrum. The magnitude of these couplings not only depends on the number and type of bonds separating the interacting pair of 1H nuclei but also on the spatial orientation between the nuclei. Both NMR chemical shifts and coupling constants provide immense information regarding structure and environment. Hence, NMR spectroscopy has become a powerful tool for the determination of organic structure.
These NMR interactions (chemical shifts and coupling constants), although very sensitive, are quite weak such that improvements in their detection have been 1 of the master goals of developments in NMR instrumentation. Such limitations are no longer severe. The use of pulses and information processing by Fourier transformation, beginning introduced past Ernst and Anderson [12] and the availability of high-field superconducting magnets have allowed for efficient indicate averaging such that nowadays, with an 11 T magnet (500 MHz), a 1H NMR spectrum can be obtained even from very dilute solutions (micromolar concentration).
Pulse Fourier transform NMR spectroscopy, equally in other spectroscopic methods, involves transitions between free energy levels. However, unlike other spectroscopic methods, the transition probability in an NMR excitation is the aforementioned regardless of chemical surroundings. NMR spectroscopy does not need to consider oscillator strengths or extinction coefficients, which are of import for infrared and UV-visible spectroscopy, respectively. The intensity of an NMR signal is determined solely by the excitation pulse, force of the external magnetic field, and temperature. The magnetic field strength and temperature determine the Boltzmann population difference between the two energy levels while the excitation pulse dictates the extent of the transition. Since only ane pulse is often used to excite all of the 1H nuclei in a sample, the extent of transitions is the same for all. Furthermore, the NMR chemic shift, which reflects the differences in resonance frequencies of inequivalent 1H nuclei, is very pocket-sized: the differences are in parts per meg (ppm). Hence, the Boltzmann distribution for the 2 spin states is substantially the aforementioned for every 1H in a molecule. Indeed, as early as 1963, the area under each tiptop in a 1H NMR spectrum has been shown to correspond proportionally to the number of hydrogen atoms sharing the same environment in a given chemical compound [13]. This quantitative aspect applies non merely to pure substances but likewise to mixtures. In fact, during the same yr, a successful quantitative analysis by 1H NMR spectroscopy of a mixture of aspirin, phenacetin, and caffeine was demonstrated [fourteen].
ii.2. 13C NMR spectroscopy
13C as well has a spin of ½ and is therefore likewise NMR active. Even so, considering the 13C isotope occurs at only 1.108%, it is difficult to detect. (The major carbon isotope, 12C, is not NMR-active.) David Grant and coworkers published a series of papers on 13C NMR spectroscopy that spanned ii decades [15, 16]. In the first paper of this series, inherent difficulties in observing 13C NMR spectra were addressed by proton decoupling and sample spinning. Since carbon atoms are frequently attached to hydrogen atoms in organic compounds, 13C-1H coupling is nowadays and leads to splitting of 13C resonances. Proton decoupling removes this interaction, consolidating multiple 13C peaks into a single taller acme. Moreover, boosted enhancement of 13C signals is observed when the 1H spin populations are perturbed, similar to the effect observed by Overhauser with electron spins [17]. Taking advantage of both the nuclear Overhauser effect (NOE) and the increased signal due to the plummet of multiple peaks, measurement of 13C NMR spectra became routine and easy to interpret. Existence in the proximity of more than one pair of electrons, 13C nuclei offer a much wider range of chemic shifts than 1H (200 ppm for 13C versus 10 ppm for 1H). In add-on, since the probability that a 13C nucleus is fastened to some other 13C nucleus is very small (about 0.0001), 13C-13C couplings are usually not observed thereby providing a much simpler 13C NMR spectrum.
Using 13C NMR spectroscopy every bit a powerful analytical tool can be easily appreciated by because the 3 isomers of a uncomplicated hydrocarbon CfiveH12 (see Figure i). n-Pentane (CHiiiCHtwoCH2CH2CH3), produces three peaks with a 1:2:ii intensity ratio, two-methylbutane ((CHiii)2CHCHiiCHiii) displays four peaks with a 1:1:2:ane intensity ratio, and neopentane ((CH3)4C) gives two peaks of 4:1 intensity ratio. For the above reasons, a qualitative and quantitative assay that is nondestructive and requires no separation is possible with 13C NMR spectroscopy [xviii]. All that one needs is a library of 13C NMR spectra of all possible components, a proficient spectral prediction software, and an efficient algorithm that tin do the search and construct a imitation spectrum that matches the observed spectrum. All of these requirements are already bachelor today. A like handling has been shown to be feasible in determining the acyl profile in various vegetable oils [19] and in characterizing the diverse sesquiterpenes in essential oils from juniper, rosemary, cedarwood, and ginger [xx].
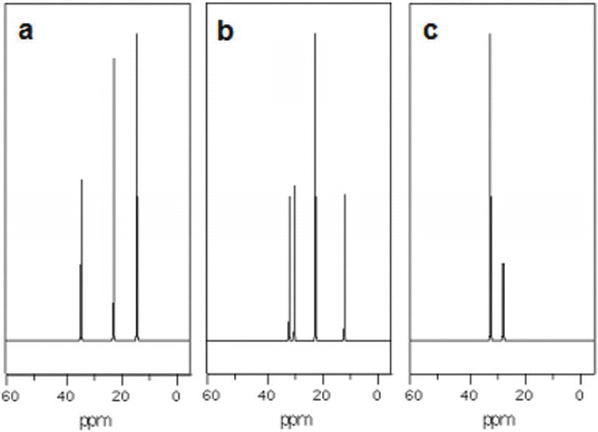
Figure 1.
13C NMR spectra of (a) n-pentane, (b) ii-methylbutane, and (c) neopentane.
The hope of a wealth of information that NMR spectroscopy offers, notwithstanding, comes also with challenges. Since the frequencies observed depend on the magnetic field strength, the peaks' shapes and widths are sensitive to the homogeneity of the magnetic field throughout the sample. Experimentally, corrections to field homogeneity are washed through a process called shimming, which involves adding small magnetic field gradients. Shimming used to be an art and both symmetry and narrowness of an NMR tiptop depended on the expertise of the NMR operator. Fortunately, with new superconducting magnets and automatic shimming, 13C NMR spectra can now be fabricated reproducible and comparable regardless of who operates the spectrometer. However, at that place are still numerous factors that are contained of the NMR operator which can affect the appearance of an NMR spectrum.
2.three. NMR chemical shifts and coupling constants
Since the frequencies (in hertz) observed for each NMR-active nucleus are dependent on the field strength, chemical shifts are reported in dimensionless units of parts per million (ppm), which then becomes independent of the magnetic field strength. Interactions betwixt nuclei, on the other hand, are independent of field strength, so these are recorded in units of frequency, hertz. Since the ppm equivalent of a hertz is determined by the strength of the magnetic field, splittings will appear narrower in a high-field magnet than in a depression-field magnet. When the coupling interactions are of the same magnitude as the chemical shift differences, the coupling blueprint is complex [21]. A hypothetical example for 1H NMR is shown in Figure two, where the coupling constant is equal to the chemical shift difference in a spectrometer operating with a 1H frequency of 100 MHz. Every bit the forcefulness of the magnetic field increases, chemical shift differences (in hertz) also increase, which can dramatically change the appearance of the spectrum. In this particular example, the spectrum only begins to announced simpler with a spectrometer operating at i GHz, in which the chemical shift difference is now 10 times bigger than the coupling constant; this is called a offset-order spectrum. Thus, 1H spectra taken at different magnetic field strengths announced dissimilar. On the other hand, 13C spectra appear similar at different magnetic field strengths. This is because 13C-13C coupling is not observed due to low natural abundance, 13C-1H couplings, although present, are always several orders of magnitude lower than the frequency difference between these 2 nuclei, and proton-decoupled 13C NMR spectra are singlets. Therefore, although 13C presents detection challenges due to its lower frequency and low natural abundance, 13C has the reward over 1H with regard to simplicity of NMR spectra.
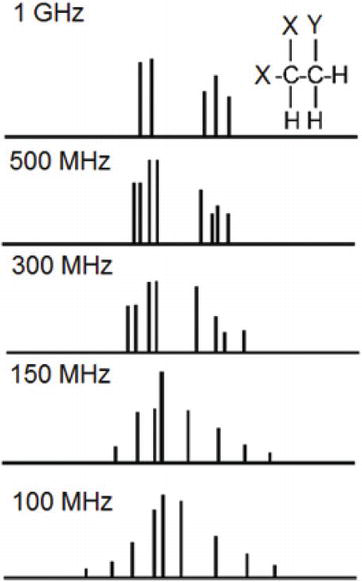
Figure 2.
1H NMR spectra of strongly coupled nuclei at various magnetic field strengths.
Accented frequencies for NMR transitions are seldom used since these numbers are dependent on the strength of the external magnetic field. Chemical shift differences are instead reported in ppm, which is the ratio of the absolute frequency with respect to the frequency of a reference compound, such equally tetramethylsilane (TMS). Alternatively, the solvent can be utilized as internal reference. Due to the sensitivity of the NMR chemical shift, intermolecular effects are also oftentimes observed [22]. Since solvents are known to induce shifts, it is important that when comparing different spectra, the same solvent should exist used. Since 13C has a much wider chemical shift range, the upshot of solvent on chemical shift is smaller for 13C (2/200) than that of 1H (0.vii/10). Furthermore, since carbon atoms, different hydrogen atoms, reside on the interior of the molecule, 13C is generally shielded from environmental furnishings, such every bit intermolecular interactions and solvent furnishings. This is one reason why 13C NMR chemical shifts are most exclusively dependent but on its covalent bonding interactions [23]. The greater susceptibility of 1H NMR chemical shifts to solvent furnishings makes 13C NMR spectroscopy a ameliorate alternative in profiling natural products. Solvent effects on both 1H and 13C NMR chemical shifts are expected to be dominated past van der Waals interactions with the solvent. These interactions are largely nonspecific thus an approximation that the solvent simply causes a abiding start on all resonances may exist valid. Using an internal reference can therefore easily remove furnishings of the medium on the observed chemic shifts. Attention, withal, is all the same required for sites that tin participate in electrostatic interactions and hydrogen bonding. 13C in carbonyl groups is one example [24].
Temperature tin too bear on observed chemical shifts through changes in the density of the sample also as changes in the internal motions of the molecule [25]. For a fair comparison of library and sample spectra, it is of import that spectra are taken at the same temperature.
Lastly, a quantitative 13C NMR spectrum requires uniform excitation of all 13C nuclei. The wider chemical shift range and lower frequency for 13C necessitate excitation pulses with much college ability with a pulse that is less than xv μs long and so that the entire chemical shift range is uniformly irradiated [26]. Furthermore, proton decoupling is as well regularly used to collapse multiple 13C peaks, but this can pb to NOEs which raise 13C nuclei that are straight leap to protons, making 13C NMR no longer uniform for carbon nuclei with different numbers of attached protons. The inverse-gated 13C NMR experiment tin exist used to overcome these problems. This involves turning the proton decoupler on only during acquisition and providing adequate time for all the 13C nuclei to relax [27]. 13C nuclei are most often relaxed past a nearby 1H nucleus. Thus, the needed relaxation time (equal to 5 ×
two.4. Reproducibility of NMR spectra
The employ of a library of NMR spectra in the analysis of a mixture of natural products requires reproducibility of both chemical shift and superlative intensity. Since samples of natural products are frequently dissolved in either dimethyl sulfoxide or methanol, confining both library and sample data to these 2 solvents tin easily ameliorate the confounding effects of the solvent on the observed chemic shifts. Modernistic NMR spectrometers are normally equipped with temperature control and so the measurements tin can be made at a given temperature, which as well avoids the temperature dependence of NMR chemical shifts, thus eliminating this trouble. The reproducibility of pinnacle intensities, yet, requires additional considerations.
The robustness of current NMR instrumentation is evident in successful indirect detection methods during which resonances from 1H nuclei spring to 12C are separated from those fastened to 13C [28]. Indirect detection is possible only if the scans or transients are highly reproducible such that these tin exist added to excerpt the desired resonances and remove completely the unwanted signals. However, this robustness only entails the reproducibility of an NMR experiment from one transient to the next. It does not address the reproducibility of NMR experiments among different laboratories. Thus, there is a need to standardize both NMR acquisition conditions and processing parameters.
The intensity of an NMR top depends on the duration of the pulse. Equalizing the m = +½ and m = −½ spin populations requires what is called in NMR spectroscopy as a xc° pulse. Peak intensities are at a maximum with this pulse. Since all nuclei in a sample are field of study to the aforementioned pulse, information technology is non necessary that a 90° pulse is always employed. For an NMR spectrum to be quantitative, the relative, not the absolute, height intensities are sufficient. Even so, the extent of the pulse determines how much time is required for relaxation betwixt transients. For signal averaging to be effective, 1 still needs to brand sure that the spins have reached equilibrium earlier applying the side by side pulse so every bit to avert saturation, which leads to loss of signal [29]. When a 90° pulse is employed, the time between transients must exist at least v times equally long as the relaxation time. The fourth dimension required between pulses can be reduced past using a smaller flip angle. For case, a 30° pulse requires a delay that is three times shorter. This reduces the top intensity for each scan but reduces the delay time required between scans enabling the conquering of more scans for the same amount of time. Another parameter that tin affect the appearance of an NMR spectrum is acquisition fourth dimension, which determines spectral resolution. What is straight acquired from an NMR experiment is a free induction decay (FID), which still needs to exist processed to produce the frequency spectrum. During processing, apodization, cipher-filling, and baseline and phase corrections are normally applied. All of these can significantly modify the integrated areas under the peaks of an NMR spectrum. Thus, a listing of universal parameters for quantitative NMR has been established past national and international round robin tests [xxx] which includes temperature (300 Thousand), pulse angle (30°), preacquisition delay (five due south), acquisition time (iii.4 s), relaxation delay (seven/iii of relaxation fourth dimension), and line broadening (0.3 Hz). For processing, careful transmission phase and baseline corrections are recommended since automated features of popular NMR processing software packages are unreliable. This validation has been performed with 1H NMR, simply these can be applied to 13C. With 13C, relaxation times are appreciably longer so relaxation agents such equally paramagnetic compounds have been used as in the earlier piece of work on petroleum distillates [31].
ii.v. Sensitivity and dynamic range
For the unbiased profiling of natural products extracts, one needs to consider the problems of sensitivity and dynamic range. A natural product extract typically contains major and small components. Oft, in order to detect minor components, it is necessary to employ separation techniques, such equally successive fractionation and chromatography which have the effect of increasing sensitivity to minor constituents and improving dynamic range. Nevertheless, this introduces bias.
Limits of detection and quantification are often given in terms of indicate to noise ratios. The International Conference on Harmonization of Technical Requirements recommends a signal to noise ratio of iii and ten for the detection limit and quantification limit, respectively (ICH Proficient Working Grouping, 1994). In practice, for fault values less than ane%, a signal to noise ratio of 150 is recommended [30]. The signal-to-dissonance ratio (South/N) in NMR spectroscopy nevertheless depends not only on concentration but also on other factors [32]:
In this equation,
Some other consideration is dynamic range. For 1H NMR, signals arising from the solvent, in particular h2o, tin can easily use up virtually of the higher $.25 in a spectrometer'south digitizer thereby decreasing the precision of signals coming from the natural product constituents. This can exist alleviated past suppressing solvent resonances, just this introduces the problem of reproducibility between runs and remains a trouble for components which have signals near the solvent.
A quantitative comparison using three magnetic field strengths—300, 400, and 500 MHz—showed that there was no difference in the sensitivity and that the standard protocol could differentiate plant samples which were spiked with 0.2 mg/mL of rutin (MW 610.5; 328 μM). This is due to the mild dependence of South/N on the field force.
For the awarding of 1H NMR for design recognition, the employ of the magnitude spectrum has been suggested [34]. The standard 1H NMR spectrum utilizes the phase-corrected existent component of the Fourier transform of the free induction decay (FID), discarding the imaginary component. This yields the absorption spectrum which is useful for normal qualitative assay due to its practiced peak resolution. Withal, this procedure sacrifices reproducibility. The use of the magnitude spectrum, which utilizes the absolute value of both the existent and imaginary components of the FID improves the reproducibility of the spectra thereby improving its accuracy for pattern recognition. This method is applicable to one-dimensional 1H NMR.
Top integrals in an NMR spectrum unfortunately are also sensitive to data processing. Apodization, zero-filling, phase and baseline corrections, and the integration itself can affect the signal-to-noise ratio of an NMR spectrum. Thus, the electric current limit in the sensitivity of NMR-based metabolomics is not due to magnetic field strength, merely is due to the current data processing methodology which uses spectral binning (alternatively called bucketing) and PCA. The usual bin size for 1H NMR is 0.04 ppm. This divides a 10 ppm 1H spectrum into 250 bins, which effectively becomes the resolution of the method. A smaller bin size can be used if the variability in the chemical shift tin can be minimized. Another trouble observed is the effect of different solvent (run across below) to move the position of chemic shifts, which will make identification using database comparisons hard [35].
2.half dozen. Event of solvent
Because institute samples incorporate a wide variety of compounds with corresponding differences in polarity, the solvent used for extraction and the NMR assay is very important. The solvent organization must remainder the power to perform a comprehensive extraction with solvent complication and reproducibility. In particular, multi-component solvent systems are prone to variation, and if there is a broad departure in vapor pressures (humid points), the solvent limerick may modify if care is not taken. Acetone and acetonitrile are effective solvents simply their use is limited by their low boiling points. The use of methanol-D4 in combination with deuterated h2o (one:1) accept been reported. By using these deuterated solvents, the extracts tin can be measured direct after extraction without need for evaporation and reconstitution. However, utilise of h2o will innovate a strong h2o elevation in the 1H NMR spectrum that must be irradiated. This becomes a source of variability around the water peak across dissimilar operators and instruments. To avoid shifts due to differences of pH in 1H NMR measurements a buffer, such as KH2PO4, is used [36].
Advertisement
3. Recent applications
NMR is capable of providing simultaneous access to both qualitative (chemic structure) and quantitative data. Unfortunately, NMR has been more generally associated with multidimensional qualitative NMR used in structural analysis and qNMR has been living under this shadow. Fan (1996) pointed out that comprehensive metabolite profiling of complex food products can exist done using i- and two-dimensional NMR assay [37]. However, it is in the apply of NMR combined with chemometric methods that the extraordinary potential of both the qualitative and quantitative applications accept been realized [38].
In view of its power to be used as an exhaustive molecular fingerprinting technique, 1H NMR has been plant to be a suitable method for the identification, quality command, and fraud detection of essential oils, a role normally reserved for GC-MS [39]. NMR fingerprinting involves obtaining 1H or 13C spectra of whole solvent extracts nether standardized conditions and ignoring, at least initially, the assignment of peaks. Multivariate statistical methods, such every bit PCA, are used to compare spectra from the samples to identify clusters and then that inferences can exist drawn about the classification of individual plant samples. The identities of metabolites responsible for differences between groups can exist investigated from loadings plots generated by PCA [40]. The following section will cover applications of 1H NMR in one- and 2-dimensions and 13C NMR together with the statistical tools.
3.1. Metabolomic profiling using 1H NMR
One-dimensional 1H NMR (1D HNMR) tin can be used in the untargeted and targeted fashion. The earliest utilize of 1D HNMR for the profiling of complex extracts had the objective of monitoring the major components of exudates of plants, such as its root system. The relative increase or decrease of primary metabolites, such every bit lactate, ethanol, and certain amino acids, could exist observed [41]. Notwithstanding, its application to natural product compounds is more than challenging due to their more complex structures and lower concentrations. Considering of its simplicity and speed, 1D HNMR in the untargeted mode tin be used past itself or equally a first-pass screening to obtain cluster and contour data using HCA and PCA [42]. The majority of HNMR studies combine 1D HNMR for PCA analysis with 2-dimensional homonuclear (1H-1H) or heteronuclear (1H-13C) NMR methods for identification of natural product metabolites.
3.ane.1. One-dimensional 1H NMR
This section discusses applications that brand use of 1D HNMR lone. The number of such studies is limited because of the presence of overlapping signals and the need for high magnetic fields. 1D HNMR at 500 MHz was used to authenticate grapes for wine making past analyzing their pare and pulp at maturity. Spectral information were reduced by binning using 0.04 ppm bin size and normalized to generate 183 variables to draw each spectrum. Chemometric methods, in particular PCA and partial least squares (PLS), enabled the identification of compounds that contributed to differences between berries, due to the sugars (glucose, fructose, and sucrose), organic acids (tartaric, malic, citric, and succinic acids), and amino acids (proline, arginine, gamma-aminobutyric acrid, valine, alanine, leucine, and isoleucine) [43].
A set up of green teas selected from a Japanese tea competition were analyzed by 1D HNMR at 750 MHz to allocate tea quality with respect to that judged by tea tasters and to propose a quality prediction model. PCA metabolomics profiling revealed a separation between the high- and the depression-quality green teas. The sense of taste mark compounds contributing to the bigotry of tea quality were identified from 1D HNMR every bit caffeine, theanine, epigallocatechin-3-gallate, epigallocatechin, epicatechin-3-gallate, and epicatechin [44].
The use of the magnitude spectrum showed good reproducibility in the analysis of 4 diverse natural product samples (12 tea extracts, eight liquor samples, 9 hops extracts, and 25 cannabis extracts) using 1D HNMR at 500-MHz and diverse statistical tools [45].
3.i.2. Ii-dimensional 1H NMR
Because of problems of bespeak overlaps in 1D HNMR spectra, two-dimensional NMR techniques are usually used to overcome these limitations. The 2d methods include 1H
1D and 2D NMR at 600 MHz together with chemometric assay were used to differentiate the origin, purity, and processing methods of chamomile flowers which were obtained from three different countries. The extracts were dissolved in DtwoO phosphate buffer adjusted to pH 7.four. 1D NMR data were analyzed by PCA assay to determine the groupings past pattern recognition and second COSY and 2nd TOCSY pulse sequences were used to assign the resonances and identify constituents [46].
Several NMR-based metabolomic studies have been done on green tea (
The same strategy was used for chemotaxonomic classification of 11 South American
1D HNMR fingerprinting followed by second TOCSY and 2D HSQC methods were used to distinguish four Asian and four Korean ginseng products, likewise as their commercial products. In this mode, the major metabolites—glutamine, arginine, sucrose, malate, and myo-inositol—were identified as chemical markers for quality balls [52]. In a study on Indian ginseng,
This NMR-based metabolomic strategy was practical to clarify seven spices used in traditional Mediterranean cuisine and to detect metabolic changes over dissimilar seasons. Both principal and secondary metabolites were identified and quantified. The major secondary metabolites identified were polyphenols, including flavonoids (apigenin, quercetin, and kaempferol derivatives) and phenylpropanoid derivatives (chlorogenic and rosmarinic acid). This written report was performed using a 300 MHz NMR musical instrument [54].
The application of NMR-based metabolomics method in plant breeding has been reported. Using a 500 MHz instrument, the NMR-based metabolomics was applied to the identification of saccharide beet (
iii.2. Metabolomic profiling using 13C NMR
Because of its lower sensitivity and longer conquering fourth dimension, 13C NMR is used less oft than 1H NMR. All the same, 13C NMR spectra are simpler, have less severe problems with overlapping peaks, are more than comparable beyond different magnetic field strengths, and are less susceptible to solvent effects. In addition, the singlet nature of 13C NMR signals makes it easier to determine the identity of individual compounds in a mixture.
13C NMR methodology was used to report the triacylglycerols of the oil extracted from the seeds of
13C NMR was used in a dereplication strategy for the identification of natural product compounds directly from institute extracts. The whole extract was first separated into fractions of simpler composition, which were and so analyzed by 13C NMR. The 13C spectra of all the fractions were aligned and subjected to design recognition by HCA. This yielded correlations among 13C signals within each fraction which were visualized equally chemic shift clusters, which were assigned to specific compounds in a 13C database. This strategy was applied to the analysis of 5 g of a bark extract from the African tree
Chemic profiling and standardization of the methanol extract from the leaves of
13C NMR is beingness used more ofttimes for dereplication of natural production extracts without fractionation. This approach is existence enhanced by availability of 13C NMR databases and predictive software which list compounds that are most likely to be present in the extract. These results have been found to be comparable to those obtained using LC-MS and GC-MS, which require fractionation and sample training [62].
The combined apply of loftier-resolution 1H and 13C NMR analysis has the potential to reveal more details that are non available using merely i technique. This combined approach was employed to detect and quantify a wide range of triacylglycerols and their component fatty acids in marine cod liver oil supplements. The combination of 1H and 13C spectra permitted the detailed analysis of components, including sn-1 monoacylglycerols, sn-1,2- and sn-1,iii-diacylglycerol adducts, and other minor components, such as trans-fat acids, free glycerol and cholesterol, and added vitamins A and E and synthetic compounds, such as ethyl docosahexaenoate or eicosopentaenoate. The identity of each compound was confirmed using 2D COSY [63].
Advertisement
4. Future prospects
The employ of 1H and 13C NMR for the profiling of natural products extracts is a rapidly growing branch of metabolomics. Information technology will further accelerate with the increasing use of NMR in quality management, the growth of NMR databases, the evolution of portable and benchtop NMR instrumentation, and advances in the use of statistical assay. Despite its considerable potential, the routine application of this method is limited by the lack of expertise to run sophisticated NMR experiments and the lack of computational tools for NMR spectral deconvolution, in particular of 1H spectra [64].
4.1. NMR in quality management
NMR has been used for the monitoring and quality management of foods, beverages, cosmetics, and pharmaceuticals. The same can be done for the profiling of natural products. In social club to ensure reproducibility and reliability and to minimize experimental artifacts, the entire process—from sample collection and storage, extraction, NMR measurement and data processing, and statistical analysis—should exist optimized and standardized [65, 66]. The NMR solvent is of detail importance because of its influence on the chemical shift positions of protons in phenolic compounds [67] and other solvent effects. This problem is more than severe for 1H every bit compared with 13C NMR.
It has been claimed that periodic calibration tin deliver accuracy every bit high as 99.9% and precision every bit adept as 0.59%, and if calibration is performed with each study, the accurateness and precision tin reach 100 and 0.35%, respectively [68]. The various experimental parameters are listed beneath:
-
Sample preparation: homogeneity of sample, extraction solvent, extraction method, and NMR solvent.
-
Conquering parameters: temperature, acquisition fourth dimension, pulse angle, number of data points, time filibuster (relaxation time), and electronic amplification.
-
NMR data processing: smoothing, stage correction, baseline correction, and indicate integration.
four.ii. NMR databases in natural products
The usefulness of NMR databases is premised on the reproducibility of the NMR experiment—starting with sample preparation, NMR acquisition, and processing—beyond unlike laboratories. It is important to avert conditions that alter the position of chemical shifts, which volition make identification using database comparisons difficult. Open-access and user-contributed 1H and 13C NMR spectral databases have a loftier potential equally a useful tool for natural products researchers provided that sample preparation, instrumentation, and acquisition parameters are standardized. For sample preparation, only selected NMR solvents should be used. Magnetic field strength is more critical for 1H than 13C NMR. Acquisition and processing parameters should exist standardized. As of 2015, 1829 1H NMR and 1383 13C NMR spectra have been available in open-access chemical databases. To further promote participation by researchers, the entire process, from information conquering, conversion of vendor-specific raw information files, and information deposition have to be simplified and standardized [69].
iv.iii. Portable and benchtop NMR instrumentation
NMR is unremarkably considered to be an expensive analytical technique which is used for research purposes but. However, for NMR to go more useful for the natural products industry where many of the companies are small to medium in size, more affordable instrumentation is needed. There have been numerous announcements regarding the development of portable and benchtop NMR instruments with full spectrum 1H and 13C NMR capability using microcoils with small portable magnets of up to ii T (approximately 85 MHz 1H) [70]. Although these are limited in capability and reproducibility compared with a full laboratory NMR musical instrument, they tin can be used in the field or production site where cryogenic liquids and stable power are not available. Considering in that location is a demand for such instrumentation for other purposes, such as forensic investigation, detection of explosives, and medical diagnostics, their evolution is certain to accelerate. This will expand the use of NMR for the profiling of natural products.
4.iv. Advances in the utilise of statistical analysis
Although the use of NMR in the analysis of biological extracts was already beingness done in the 1980s, information technology was the application of statistical methods that enabled researchers to make apply the big amount of NMR information to detect patterns and correlations. The first step unremarkably involves the simplification of large NMR data sets to discover relationships, groupings, or dependencies using PCA. Second, the groups can be classified with or without a training gear up which has known information or characteristics confronting which other sample sets are compared. Linear discriminant analysis (LDA) and soft independent modeling of class illustration (SIMCA) are used for this purpose. For quantitative analysis of constituents, in detail for strongly overlapping peaks, principal component regression (PCR) or PLS regression can be used [71]. Although these statistical techniques are now commonly used, new ones go along to be developed and reported.
One of the near exciting areas of development is the use of statistical methods to correlate NMR signals with biological activeness. Since the NMR signals can be related to specific compounds, this in effect allows 1 to correlate specific compounds with biological activeness. Although information technology has to exist emphasized that correlation is non proof of biological activeness, this strategy notwithstanding allows one to shortcut the procedure of discovering bioactive compounds in a complex natural product mixture. This also allows one to find multiple active compounds.
Advertizing
five. Conclusions
1H and 13C NMR are rapidly expanding it office from its traditional use mainly every bit a qualitative spectroscopic technique for the determination of chemical structure to a quantitative tool for the metabolomic written report of natural production extracts, whether for quality control of phytomedicine products, analysis of the metabolome for establish profiling, identification of constituents as plant markers, or for plant biotechnology. A major enabler for the utilize of NMR for metabolomic studies is the application of various statistical techniques which are able to find patterns and correlations in the big NMR data sets. The continued expansion of the use of NMR for the metabolomic profiling of natural production extracts volition probable depend on the further development of statistical methods and the availability of NMR databases for both 1H and 13C nuclei. It is likely that more than compounds will exist identified as techniques are improved.
An NMR spectrum is quantitative. An agreement of the physical principles of NMR provides the theoretical basis for its apply as a quantitative tool. NMR spectroscopy does non require a standard for each component since the intensity of each point is directly proportional to the number of nuclei being observed regardless of environment. NMR spectroscopy likewise offers detailed information regarding molecular structure. Using NMR spectroscopy as a tool in the profiling of natural product extracts therefore non but provides accurate and precise composition, but too structural evidence for each of the components. Since the NMR bespeak dependence on diverse factors is already well known, resonance positions and intensities are highly reproducible. These are important characteristics which give NMR a unique advantage over other analytical methods.
Advertizement
Abbreviations
| 1D HNMR | One-dimensional 1H NMR |
| 2D COSY | Two-dimensional 1H-1H correlation spectroscopy |
| 2D JNMR | 2-dimensional 1H J-resolved spectroscopy |
| 2D HMBC | Two-dimensional 1H-13C heteronuclear multibond coherence |
| 2D HSQC | Two-dimensional, 1H-13C single quantum coherence |
| 2D TOCSY | Two-dimensional total correlation spectroscopy |
| GC-MS | Gas chromatography-mass spectrometry |
| HCA | Hierarchical cluster analysis |
| LC-MS | Liquid chromatography-mass spectrometry |
| MS | Mass spectrometry |
| NMR | Nuclear magnetic resonance |
| PCA | Chief components analysis |
| PLS | Partial least squares |
| PLS-DA | Fractional least squares-discriminant analysis |
| qNMR | Quantitative NMR |
| qHNMR | Quantitative proton (1H) NMR |
| qTLC | Quantitative thin layer chromatography |
References
- 1.
Metabolomics Society. Metabolomics. [Cited 14-09-2014]. Bachelor from: http://www.metabolomicssociety.org/ - 2.
Commisso Yard, Strazzer P, Toffal K, Stocchero M, Guzzo F. Untargeted metabolomics: An emerging approach to make up one's mind the composition of herbal products. Computational and Structural Biotechnology Journal. 2013; 4 (five):e201301007. Available from:http://dx.doi.org/x.5936/csbj.201301007 - iii.
Wishart DS. Quantitative metabolomics using NMR. Trends in Analytical Chemistry. 2008; 27 (three):228-237 - 4.
Markley JL, Bruschweiler R, Edison AS, Eghbalnia 60 minutes, Powers R, Raftery D, Wishart DS. The future of NMR-based metabolomics. Current Opinion in Biotechnology. 2017; 43 :34-40 - 5.
Malz F, Jancke H. Validation of quantitative NMR. Journal of Pharmaceutical and Biomedical Assay. 2005; 38 :813-823 - 6.
Webster GK, Kumar S. Expanding the belittling toolbox: Pharmaceutical awarding of quantitative NMR. Analytical Chemistry. 2014; 86 :11474-11480 - seven.
Simmler C, Kulakowski D, Lankin DC, McAlpine JB, Chen SN, Pauli GF. Holistic analysis enhances the description of metabolic complication in dietary natural products. Advances in Nutrition. 2016; vii :179-189 - viii.
Wishart DS. Quantitative metabolomics using NMR. Trends in Analytical Chemistry. 2008; 27 (3):228-237 - ix.
Wold S. Chemometrics; what exercise nosotros hateful with it, and what do we want from information technology? Chemometrics and Intelligent Laboratory Systems. 1995; thirty :109-115 - 10.
Bansal A, Chhabra Five, Rawal RK, Sharma S. Chemometrics: A new scenario in herbal drug standardization. Journal of Pharmaceutical Assay. 2014; 4 (4):223-233 - eleven.
Kim HK, Saifullah KS, Wilson EG, Prat Kricun SD, Meissner A, Goraler Due south, Deelder AM, Choi YH, Verpoorte R. Metabolic nomenclature of S American Ilex species by NMR-based metabolomics. Phytochemistry. 2010; 71 :773-784 - 12.
Ernst RR, Anderson WA. Application of Fourier transform spectroscopy to magnetic resonance. The Review of Scientific Instruments. 1966; 37 :93-102 - 13.
Jungnickel JL, Forbes JW. Quantitative measurement of hydrogen types by integrated nuclear magnetic resonance intensities. Belittling Chemistry. 1963; 35 :938-942 - xiv.
Hollis DP. Quantitative analysis of aspirin, phenacetin, and caffeine mixtures past nuclear magnetic resonance spectrometry. Analytical Chemistry. 1963; 35 :1682-1684 - 15.
Paul EG, Grant DM. Carbon-13 magnetic resonance. I. Improved carbon-13 magnetic resonance spectra obtained past proton decoupling and rapid sample spinning. Periodical of the American Chemic Society. 1964; 86 :2977-2983 - sixteen.
Dalling DK, Ladner KH, Grant DM, Woolfenden WR. Carbon-13 magnetic resonance. 27. The dependence of chemical shifts on methyl rotational conformations and dynamics in the methylated benzenes and naphthalenes. Journal of the American Chemical Club. 1977; 99 :7142-7150 - 17.
Overhauser AW. Polarization of nuclei in metals. Physics Review. 1953; 92 :411-415 - xviii.
Laude DA, Wilkins CL. Identification of organic mixture components without separation: Quantitative and edited carbon-13 nuclear magnetic resonance spectrometry information for analysis of petroleum distillates. Analytical Chemistry. 1986; 58 :2820-2824 - nineteen.
Wollenberg KF, Kurt F. Quantitative loftier resolution 13C nuclear magnetic resonance of the olefinic and carbonyl carbons of edible vegetable oils. Journal of the American Oil Chemists' Society. 1990; 67 :487-494 - xx.
Tomi F, Bradesi P, Bighelli A, Casanova J. Computer-aided identification of individual components of essential oils using carbon-13 NMR spectroscopy. Periodical of Magnetic Resonance Assay. 1995; 1 :25-34 - 21.
Swalen JD, Reilly CA. Analysis of circuitous NMR spectra. An iterative method. The Periodical of Chemic Physics. 1962; one :21-29 - 22.
Jameson CJ. Gas-phase NMR spectroscopy. Chemical Reviews. 1991; 91 :1375-1395 - 23.
De Dios Air-conditioning, Pearson JG, Oldfield E. Secondary and tertiary structural effects on protein NMR chemic shifts: An ab initio approach. Scientific discipline. 1993; 260 :1491-1496 - 24.
De Dios Air conditioning, Oldfield East. Chemic shifts of carbonyl carbons in peptides and proteins. Journal of the American Chemical Society. 1994; 116 :11485-11488 - 25.
De Dios AC, Jameson CJ. The NMR chemical shift: Insight into structure and environment. Annual Reports on NMR Spectroscopy. 1994; 29 :1-69 - 26.
Skinner TE, Reiss TO, Luy B, Khaneja N, Glaser SJ. Application of optimal command theory to the pattern of broadband excitation pulses for high-resolution NMR. Periodical of Magnetic Resonance. 2003; 163 :8-15 - 27.
Freeman RH, Hill DW, Kaptein R. Proton-decoupled NMR. Spectra of carbon-13 with the nuclear Overhauser effect suppressed. Journal of Magnetic Resonance. 1972; 7 :327-329 - 28.
Bax A, Subramanian Due south. Sensitivity-enhanced two-dimensional heteronuclear shift correlation NMR spectroscopy. Journal of Magnetic Resonance. 1986; 67 :565-569 - 29.
Ernst RR, Anderson WA. Application of Fourier transform spectroscopy to magnetic resonance. The Review of Scientific Instruments. 1966; 37 :93-102 - thirty.
Malz F, Jancke H. Validation of quantitative NMR. Journal of Pharmaceutical and Biomedical Analysis. 2005; 38 :813-823 - 31.
Laude DA, Wilkins CL. Identification of organic mixture components without separation: Quantitative and edited carbon-13 nuclear magnetic resonance spectrometry data for analysis of petroleum distillates. Analytical Chemistry. 1986; 58 :2820-2824 - 32.
Bharti SK, Roy R. Quantitative 1H NMR spectroscopy. Trends in Analytical Chemical science. 2012; 35 :5-26 - 33.
Hsieh PY, Widegren JA, Slifka AJ, Hagen AJ, Rorrer RA. Direct measurement of trace polycyclic aromatic hydrocarbons in diesel with 1H and 13C NMR spectroscopy: Event of PAH content on fuel lubricity. Free energy and Fuels. 2015; 29 (seven):4289-4297 - 34.
Harrington PB, Wang X. Spectral Representation of proton NMR spectroscopy for the design recognition of complex materials, Published online: 24 February 2017. Journal of Analysis and Testing. 2017. DOI: x.1007/s41664-017-0003-y - 35.
Van der Kooy F, Venkataya B, Pearson JL, Torres A, Li CG, Chang D. Sensitivity of NMR-based metabolomics in drug discovery from medicinal plants. European Journal of Medicinal Plants. 2015; five (2):191-203 - 36.
Verpoorte R, Choi YH, Kim HK. NMR-based metabolomics at piece of work in phytochemistry. Phytochemistry Reviews. 2007; half dozen :3-14 - 37.
Fan TWM. Metabolite profiling by 1- and ii-dimensional NMR assay of circuitous mixtures. Progress in Nuclear Magnetic Resonance Spectroscopy. 1996; 28 :161-219 - 38.
Pauli GF, Jaki BU, Lankin DC. Quantitative 1H NMR: Development and potential of a method for natural products analysis. Journal of Natural Products. 2005; 68 :133-149 - 39.
Guerrini A, Sacchetti G, Muzzoli M, Rueda GM, Medici A, Besco Eastward, Bruni R. Composition of the volatile fraction of Ocotea bofo Kunth (Lauraceae) calyces past GC-MS and NMR fingerprinting and its antimicrobial and antioxidant activity. Journal of Agricultural and Food Chemistry. 2006;54 :7778-7788 - 40.
Ward JL, Baker JM, Beale MH. Recent applications of NMR spectroscopy in plant metabolomics. The FEBS Journal. 2007; 274 :1126-1131 - 41.
Fan TWM, Higashi RM, Lane AN. Monitoring of hypoxic metabolism in superfused found tissues past in vivo 1H NMR. Archives of Biochemistry and Biophysics. 1986; 251 (two):674-687 - 42.
PSimmler C, Napolitano JG, McAlpine JB, Chen SN, Pauli GF. Universal quantitative NMR analysis of circuitous natural samples. Electric current Opinion in Biotechnology. 2014. DOI: 10.1016/j.copbio.2013.08.004 - 43.
Pereira GE, Gaudillere JP, van Leeuwen C, Hilbert Yard, Maucourt Yard, Deborde C, Moing A, Rolin D. 1H NMR metabolite fingerprints of grape drupe: Comparison of vintage and soil effects in Bordeaux grapevine growing areas. Analytica Chimica Acta. 2006; 563 :346-352 - 44.
Tarachiwin 50, Ute K, Kobayashi A, Fukusaki E. 1H NMR based metabolic profiling in the evaluation of Japanese light-green tea quality. Journal of Agricultural and Food Chemical science. 2007; 55 :9330-9336 - 45.
Harrington PB, Wang X. Spectral Representation of proton NMR spectroscopy for the pattern recognition of complex materials, Published online: 24 Feb 2017. Journal of Analysis and Testing. 2017. DOI: 10.1007/s41664-017-0003-y - 46.
Wang Y, Tang H, Nicholson JK, Hylands PJ, Sampson J, Whitcombe I, Stewart CG, Caiger S, Oru I, Holmes E. Metabolomic strategy for the classification and quality command of phytomedicine: A case report of chamomile flower ( Matricaria recutita L.). Planta Medica. 2004;seventy :250-255 - 47.
Le Gall G, Colquhoun IJ, Defernez Thou. Metabolite profiling using 1H NMR spectroscopy for quality assessment of dark-green tea, Camellia sinensis (L.). Journal of Agronomical and Nutrient Chemistry. 2004;52 :692-700 - 48.
Lee JE, Lee BJ, Chung JO, Hwang JA, Lee SJ, Lee CH, Hong YS. Geographical and climatic dependencies of green tea ( Camellia sinensis ) metabolites: A 1H NMR-based metabolomics study. Journal of Agricultural and Food Chemistry. 2010;58 :10582-10589 - 49.
Lee JE, Lee BJ, Hwang JA, Ko KS, Chung JO, Kim EH, Lee SJ, Hong YS. Metabolic dependence of green tea on plucking positions revisited: A Metabolomic written report. Periodical of Agricultural and Food Chemistry. 2011; 59 :10579-10585 - 50.
Wahyuni DSC, Kristanti MW, Putri RK, Rinanto T. NMR metabolic profiling of light-green tea ( Camellia sinensis 50.) leaves grown at Kemuning, Republic of indonesia. IOP Conference Series: Journal of Physics: Conference Series. 2017;795 :012013 - 51.
Kim HK, Saifullah KS, Wilson EG, Prat Kricun SD, Meissner A, Goraler S, Deelder AM, Choi YH, Verpoorte R. Metabolic nomenclature of South American Ilex species by NMR-based metabolomics. Phytochemistry. 2010;71 :773-784 - 52.
Lee EJ, Shaykhutdinov R, Weljie AM, Vogel HJ, Facchini PJ, Park SU, Kim YK, Yang TJ. Quality assessment of ginseng past 1H NMR metabolite fingerprinting and profiling assay. Journal of Agricultural and Food Chemistry. 2009; 57 :7513-7522 - 53.
Namdeo AG, Sharma A, Yadav KN, Gawande R, Mahadik KR, Lopez-Gresa MP, Kim HK, Choi YH, Verpoorte R. Metabolic label of Withania somnifera from dissimilar regions of Republic of india using NMR spectroscopy. Planta Medica. 2011;77 (17):1958-1964 - 54.
Scognamiglio M, D'Abrosca B, Esposito A, Fiorentino A. Chemical limerick and seasonality of effluvious mediterranean constitute species by NMR-based metabolomics. Journal of Analytical Methods in Chemistry. 2015 Article ID 258570, 9 pages. http://dx.doi.org/ten.1155/2015/258570 - 55.
Sekiyama Y, Okazaki K, Kikuchi J, Ikeda Southward. NMR-based metabolic profiling of fieldgrown leaves from sugar beet plants Harbouring unlike levels of resistance to Cercospora foliage spot affliction. Metabolites. 2017;seven :(4). 13 pages. DOI: 10.3390/metabo7010004 - 56.
Vlahov One thousand, Chepkwony PK, Ndalut PK. 13C NMR characterization of triacylglycerols of moringa oleifera seed oil: An "oleic-Vaccenic acid" oil. Periodical of Agronomical and Food Chemical science. 2002; 50 :970-975 - 57.
Standal IB, Axelson DE, Aursand Thousand. Authentication of marine oils using 13C NMR spectroscopy. Lipid Technology. 2011; 23 (7):152-154 - 58.
Aursand Yard, Standal IB, Prael A, McEvoy L, Irvine J, Axelson DE. 13C NMR pattern recognition techniques for the nomenclature of Atlantic Salmon ( Salmo salar L.) according to their wild, farmed, and geographical origin. Periodical of Agricultural and Food Chemistry. 2009;57 :3444-3451 - 59.
Aursand K, Standal IB, Axelson DE. High resolution 13C nuclear magnetic resonance spectroscopy blueprint recognition of fish oil capsules. Periodical of Agricultural and Food Chemistry. 2007; 55 :38-47 - 60.
Hubert J, Nuzillard JM, Purson S, Hamzaoui M, Borie North, Reynaud R, Renault JH. Identification of natural metabolites in mixture: A design recognition strategy based on 13C NMR. Analytical Chemistry. 2014; 86 :2955-2962 - 61.
Lagurin LG, Magsalin JDJ, Zosa AR, Dayrit FM. Chemical profiling and chemical standardization of Vitex negundo using 13C NMR. Journal of Medicinal Plants Enquiry. 2017;11 (1):11-21 - 62.
Bakiri A, Hubert J, Reynaud R, Lanthony Southward, Harakat D, Renault JH, Nuzillard JM. Estimator-aided 13C NMR chemic profiling of rough natural extracts without fractionation. Journal of Natural Products 2017; 80 :1387-96 - 63.
Siddiqui North, Sim J, Silwood CJL, Toms H, Iles RA, Grootveld M. Multicomponent assay of encapsulated marine oil supplements using high-resolution 1H and 13C NMR techniques. Journal of Lipid Research. 2003; 44 :2406-2427 - 64.
Mahrous EA, Farag MA. Two dimensional NMR spectroscopic approaches for exploring found metabolome: A review. Journal of Advanced Research. 2015; 6 :3-15 - 65.
Rivas-Ubach A, Perez-Trujillo M, Sardans J, Gargallo-Garriga A, Parella T, Penuelas J. Ecometabolomics: Optimized NMR-based method. Methods in Ecology and Development. 2013; four :464-473 - 66.
Schönberger T, Monakhova YB, Lachenmeier DW, Kuballa T, the Non-Profit Good Team (Next)-NMR working group Germany. Guide to NMR method evolution and validation – Function 1: Identification and quantification. In: EUROLAB Technical Study 1/2014 - 67.
Kim HK, Choi YH, Verpoorte R. NMR-based metabolomic analysis of plants. Nature Protocols. 2010; five :536-549 - 68.
Crouch R, Russell D. Easy, Precise and Accurate Quantitative NMR. Agilent Technologies Awarding Note. 2011. [Cited 14-09-2014] http://world wide web.agilent.com - 69.
Johnson SR, Lange BM. Open-admission metabolomics databases for natural production research: Present capabilities and futurity potential. Frontiers in Bioengineering and Biotechnology. 2015; three (22):10 - lxx.
Herberg JL, Demas Five, Malba V, Bernhardt A, Evans L, Harvey C, Chinn S, Maxwell R, Reimer J, Pines A. Portable, low-cost NMR with laser-lathe lithography produced microcoils. Journal of Magnetic Resonance. 2007; 189 :121-129 - 71.
Monakhova YB, Kuballa T, Lachenmeier DW. Chemometric methods in NMR spectroscopic analysis of food products. Journal of Analytical Chemistry. 2013; 68 (9):755-766
Submitted: June 2nd, 2017 Reviewed: September 18th, 2017 Published: Dec sixth, 2017
© 2017 The Author(s). Licensee IntechOpen. This chapter is distributed nether the terms of the Creative Commons Attribution 3.0 License, which permits unrestricted use, distribution, and reproduction in whatsoever medium, provided the original work is properly cited.

Source: https://www.intechopen.com/chapters/57387
Posted by: johansenwhang1968.blogspot.com
0 Response to "What Is The Difference Between A Regular 13c Nmr And A 13c Nmr Registered In A Proton Coupled Mode?"
Post a Comment